Loss of the hepatic protein hepcidin can lead to severe iron overload with symptoms resembling those of hemochromatosis. Which of the following functions of hepcidin accounts for the iron overload when the protein is deficient?
A. activates the expression of the ironresponse element-binding protein that regulates transferring receptor and ferritin mRNA translation
B. decreases the level of intestinal membrane iron transporters, resulting in reduced iron uptake
C. facilitation of the interaction of transferring with the transferrin receptor
D. forms a complex with ferritin allowing for higher intracellular storage
E. promotes the formation of hemosiderin, thus detoxifying iron
Correct Answer: B
Section: Biochemistry Hepcidin is a hepatically synthesized iron regulatory protein that functions by inhibiting the presentation of one or more of the iron transporters [e.g., DMT1 and Ireg1 (ferroportin)] in intestinal membranes. With a high iron diet, the level of hepcidin mRNA increases and conversely its levels decrease when dietary iron is low. This is occurring simultaneous to reciprocal changes in the levels of the transporters proteins themselves. Loss of hepcidin activity would then lead to unregulated iron uptake from the intestines leading to iron overload. In fact, it is now considered that defects in hepcidin function contribute to the development of hemochromatosis. Hepcidin does not activate expression of iron-response elementbinding protein (choice A), facilitate transferring interaction with the transferring receptor (choice C), form a complex with ferritin (choice D), nor promote the formation of hemosiderin (choice E).
Question 532:
In an enzyme with a critical Glu residue in the active site, which of the following amino acid substitutions would be expected to have the least effect on enzyme activity?
A. Arg
B. Asp
C. Lys
D. Ser
E. Tyr
Correct Answer: B
Section: Biochemistry Glutamic acid is an acidic amino acid at physiologic pH and therefore, substitution for another acidic amino acid, such as aspartic acid, would be expected to have minimal effect on the activity of the enzyme. Arginine (choice A) and lysine (choice C) are both basic amino acids at physiologic pH and would not be able to substitute for an acidic amino acid. Serine (choice D) and tyrosine (choice E) both have ionizable hydroxyl groups but the pKa values of those hydroxyl groups would not favor substitution for an acidic amino acid.
Question 533:
Many proteins undergo modifications during and/or following translation. The ATPdependent polyubiquitination of proteins is a signal for which of the following events?
A. addition of the oligosaccharide core to the N-linkage site adjacent to the ubiquitin
B. recognition by the toxin of diphtheria
C. recycling of the protein back to the endoplasmic reticulum (ER) from the cis-Golgi to allow proper folding
D. targeting the protein for degradation in the proteosome
E. translocation of the protein into the nucleus
Correct Answer: D
Section: Biochemistry The ATP-dependent poly-ubiquitination of proteins is a signal that the protein is misfolded and thus, needs to be targeted by the proteosome for degradation. Mono-ubiquitination is known to initiate cell signaling by allowing other proteins that contain ubiquitin-binding domains to interact with the mono- ubiquitinated substrate. Mono-ubiquitination has also been associated with targeting of membrane proteins to the lysosome. None of the other examples (choices AC, E) are associated with addition of ubiquitin.
Question 534:
A30-month-old child presents with coarse facial features, corneal clouding, hepatosplenomegaly, and exhibiting disproportionate short-trunk dwarfism. Radiographic analysis indicates enlargement of the diaphyses of the long bones and irregular metaphyses, along with poorly developed epiphyseal centers. Other skeletal abnormalities typify the features comprising dystosis multiplex. The child's physical stature and the analysis of bone development indicate the child is suffering from which of the following disorders?
A. Hunter syndrome
B. Hurler syndrome
C. Maroteaux-Lamy syndrome
D. Morquio syndrome type B
E. Sanfilippo disease type A
Correct Answer: B
Section: Biochemistry Although multiorgan involvement, liver and spleen enlargement, and skeletal abnormalities are common to all the mucopolysaccharidotic (MPS) diseases, each encompasses features that allow for specific diagnosis. Hurler syndrome is characterized by progressive multiorgan failure and premature death. Hallmark features include enlargement of the spleen and liver, severe skeletal deformity, and coarse facial features (which are associated with the constellation of defects referred to as dystosis multiplex). The disease results from a defect in alpha-L-iduronidase activity, which leads to intracellular accumulations of heparin sulfates and dermatan sulfates. The accumulation of these GAGs (glycosaminoglycan) in Hurler syndrome patients severely affect development of the skeletal system leading, primarily, to defective long bone growth plate disruption. Hunter syndrome (choice A) hasfeatures similar to that of Hurler with a lack of corneal clouding. Additionally, symptoms progress slower, with onset of symptoms occurring between 2 and 4 years of age. Maroteaux-Lamy syndrome (choice C) encompasses symptoms similar to Hurler but with normal mental development. Morquio syndrome (choice D) comprises two related disorders, both of which are characterized by short-trunk dwarfism, fine corneal deposits, and a skeletal dysplasia (spondyloepiphyseal) distinct from other MPS. Sanfilippo syndrome (choice E) comprises four recognized types characterized by severe CNS degeneration with only mild involvement of other organ systems. Symptoms do not appear until 26 years of age.
Question 535:
I-cell disease (also identified as mucolipidosis type II) is characterized by the presence of inclusion bodies in fibroblasts (hence the derivation of the term I-cell), severe psychomotor retardation, corneal clouding, and dystosis multiplex. These symptoms arise from a defect in the targeting of lysosomal enzymes due to an inability to carry out which of the following processes?
A. produce mannose-6-phosphate modifications in lysosomal enzymes
B. recycle the lysosomal receptor for mannose-6-phosphate present on lysosomal enzymes
C. remove mannose-6-phosphates from lysosomal enzymes prior to their transport to the lysosomes
D. synthesize the mannose-6-phosphate receptor found in lysosomes
E. transport mannose-6-phosphate receptors to lysosomes
Correct Answer: A
Section: Biochemistry Enzymes that are destined for the lysosomes (lysosomal enzymes) are directed there by a specific carbohydrate modification. During transit through the Golgi apparatus a residue of Nacetylglucosamine- 1phosphate is added to carbon 6 of one or more specific mannose residues that have been incorporated into these enzymes. The N-acetylglucosamine is activated by coupling to UDP and is transferred by an Nacetylglucosamine phosphotransferase yielding N-acetylglucosamine-1- phosphate-6-mannoseprotein. A second reaction removes the Nacetylglucosamine leaving mannose residues phosphorylated in the sixth position. Aspecific mannose-6-phosphate receptor is present in the membranes of the Golgi apparatus. Binding of mannose-6-phosphate to this receptor targets proteins to the lysosomes. Defects in the proper targeting of glycoproteins to the lysosomes can also lead to clinical complications. Deficiencies in Nacetylglucosamine phosphotransferase lead to the formation of dense inclusion bodies in fibroblasts. Two disorders related to deficiencies in the targeting of lysosomal enzymes are termed I-cell disease (mucolipidosis II) and pseudo-Hurler polydystrophy (mucolipidosis III). I-cell disease is characterized by severe psychomotor retardation, skeletal abnormalities, coarse facial features, painful restricted joint movement, and early mortality. Pseudo- Hurler polydystrophy is less severe; it progresses more slowly, and afflicted individuals live to adulthood. Each of the other choices (B, C, D, and E) represent other potential pathways that are not affected in the processing, delivery, or presentation of lysosomal enzymes or the receptors that recognize the properly processed enzymes.
Question 536:
A 23-year-old man sees his physician to ask about the recent appearance of several large closely spaced bumps on his elbows. Suspecting that these are fatty eruptions, the physician tests the man's blood for lipid, cholesterol, and lipoprotein levels. Results show elevated cholesterol and triglycerides and the presence of a variant form of very low-density lipoprotein (VLDL) identified as beta-migrating VLDL (VLDL). Amore careful analysis of the biochemical properties of the apoproteins associated with the beta-VLDL particles identifies a form of apo E that has a more negative charge than apo E from normal individuals. These results indicate the individual is afflicted with which of the following hyperlipoproteinemias?
A. type I (familial LPL deficiency)
B. type II (FH)
C. type III (dysbetalipoproteinemia)
D. type IV (familial hypertriglycerolemia)
E. Wolman disease
Correct Answer: C
Section: Biochemistry Familial dysbetalipoproteinemia (type III hyperlipoproteinemia) results from a genetic variant in the apo E gene that causes poor interaction of chylomicron remnants and VLDLs with the apo E receptor. This results in the presence, in the serum, of beta-migrating VLDL (- VLDLs), which are cholesterol- rich remnants of both intestinal chylomicrons and hepatic VLDL. Diagnosis of type III hyperlipoproteinemia is indicated by elevated plasma cholesterol and triglyceride, xanthomas (fatty eruptions under the skin), and of course the presence of -VLDL. Type I hyperlipoproteinemia (choice A) results from defects in the activity or activation of LPL and results in the massive accumulation of chylomicrons in the plasma. The disease is usually detected in childhood following recurrent attacks of abdominal pain, hepatosplenomegaly, and pancreatitis. Familial hypercholesterolemia (choice B) is the result of defects in the LDL receptor. The defects lead to characteristic elevation in LDL, deposition of LDL-derived cholesterol in the tendons and skin and in the arteries. Individuals homozygous for defective LDL receptors have severe hypercholesterolemia (6501000 mg/dL) and coronary heart disease begins early in childhood with death caused by myocardial infarct before the age of 20. Type IV hyperlipoproteinemia (choice D) is associated with overproduction VLDLs. An associated glucose intolerance and hyperinsulinemia are also seen in this disorder. Wolman disease (choice E) is caused by a deficiency in lysosomal acid lipase and results in massive accumulation of cholesteryl esters and triglycerides in most tissues. The disease is almost always fatal before the age of 1 year.
Question 537:
A 28-year-old man has the following symptoms: diffuse grayish corneal opacities, anemia, proteinuria, and hyperlipemia. Renal function is normal and serum albumin level is only slightly elevated. Plasma triglycerides and unesterified cholesterol levels are elevated, as are levels of phosphatidylcholine. These symptoms are indicative of which lipoproteinassociated disorder?
A. Bassen-Kornzweig syndrome
B. familial hypercholesterolemia (FH)
C. familial hypertriacylglycerolemia
D. familial lecithin-cholesterol acyltransferase (LCAT) deficiency
E. Wolman disease
Correct Answer: D
Section: Biochemistry Two familial syndromes directly involve defects in LCAT. Familial LCAT deficiency is characterized by near complete absence of the enzyme activity from the plasma. Fish eye disease is characterized by an absence of LCAT activity toward high-density lipoproteins (HDLs) but presence of activity toward LDLs. Clinical features of familial LCAT deficiency include corneal opacities, anemia, and proteinuria. Due to the lack of LCAT activity, the plasma level of esterified cholesterol is lower than normal and phosphatidylcholine (the principal source of fatty acid for esterification to cholesterol) levels are higher than normal. The profile of all classes of plasma lipoproteins in patients with familial LCAT deficiency is abnormal. Bassen-Kornzweig syndrome (choice A), also identified as abetalipoproteinemia, is due to a defect in apo B expression. Clinical symptoms include retinitis pigmentosa, ataxic neuropathy, and erythrocytes that appear thorny (acanthocytosis). FH (choice B) is characterized by reduced LDL clearance, which leads to severe hypercholesterolemia. Major clinical symptoms are arterial deposition of LDL-cholesterol, which leads to atherosclerosis and coronary artery disease. Deposition of LDL-cholesterol is also seen in tendons and skin resulting in xanthomas. Familial hypertriacylglycerolemia (choice C), also identified as hyperlipoproteinemia type IV, is a form of LPL deficiency. The defect leads to increased levels of circulating VLDLs and is associated with glucose intolerance and hyperinsulinemia. This disorder is frequently associated with Type II diabetes. Wolman disease (choice E) is cholesterol ester storage disease that leads to massive accumulation of cholesteryl esters and triglycerides in most tissues. This disease is almost always fatal within the first year of life and thus would not be present in a 28-year- old.
Question 538:
Following a minor respiratory illness, a seemingly healthy, developmentally normal 15-month-old boy exhibited repeated episodes of severe lethargy and vomiting following periods of fasting, such as during the middle of the night. The parents brought the infant to the emergency room following a seizure. The child was hypoglycemic and was administered 10% dextrose, but remained lethargic. Blood ammonia was high, liver function tests were slightly elevated, and his serum contained an accumulation of dicarboxylic acids. Only low levels of ketones were detecteable in the urine. This infant suffers from which of the following disorders?
A. glutaric acidemia type II
B. Lesch-Nyhan syndrome
C. MCAD deficiency
D. pyruvate dehydrogenase (PDH) deficiency
E. type III (Cori) glycogen storage disease
Correct Answer: C
Section: Biochemistry Deficiency in MCAD is the most common inherited defect in the pathways of mitochondrial fatty acid oxidation. The most common presentation of infants with this disorder is episodic hypoketotic hypoglycemia following periods of fasting. Although the first episode may be fatal, and incorrectly ascribed to sudden infant death syndrome, patients with MCAD deficiency are normal between episodes and are treated by avoidance of fasting and treatment of acute episodes with intravenous glucose. Accumulation of acylcarnitines (dicarboxylic acids) is diagnostic, in particular octanoylcarnitine. Glutaric acidemia type II (choice A) results from a defect in electron transfer flavoproteinubiquinone oxidoreductase and presents with symptoms of hypoketotic hypoglycemia as in the case of MCAD deficiency. However, this disorder manifests within the first 2448 h after birth and is frequently associated with congenital anomalies. Lesch-Nyhan syndrome (choice B) results from a defect in HGPRT--an enzyme involved in nucleotide metabolism. Symptoms of Lesch-Nyhan syndrome include hyperuricemia, bizarre neurobehavioral manifestations, growth retardation, and anemia. Deficiency in PDH (choice D) results in lactic acidemia, which can be quite severe at birth leading to neonatal fatality. Milder deficiency results in lactic acidemia associated with profound psychmotor retardation. Cori disease (choice E) results from a defect in the glycogen debranching enzymes. Clinical features include hepatomegaly, hypoglycemia, skeletal myopathy and short stature, and cardiomyopathy.
Question 539:
Infants exhibiting profound metabolic ketoacidosis, muscular hypotonia, developmental retardation, and who have very large accumulations of methylmalonic acid in their blood and urine suffer from a disorder known as methylmalonic acidemia. This disorder results from a defect in which of the following enzymes?
A. alpha-keto acid dehydrogenase
B. homogentisic acid oxidase
C. methylmalonyl-CoA mutase
D. phenylalanine hydroxylase
E. tyrosine aminotransferase
Correct Answer: C
Section: Biochemistry Defects in methylmalonyl-CoA mutase activity comprise four distinct genotypes whose clinical symptoms are remarkably similar. Characteristic findings in methylmalonyl-CoA mutase deficiency include failure to thrive leading to developmental abnormalities, recurrent vomiting, respiratory distress, hepatomegaly, and muscular hypotonia. In addition, patients have severely elevated levels of methylmalonic acid in the blood and urine. Unaffected individuals have near undetectable levels of methylmalonate in their plasma, whereas, affected individuals have been found to have levels ranging from 3 to 40 mg/dL in their blood. Deficiency in alpha-ketoacid dehydrogenase (choice A) results in MSUD, so named because of the characteristic odor of the urine in afflicted individuals. Mental retardation in the MSUD is extensive. Deficiency in homogentisic acid oxidase (choice B) results in alkaptonuria. Alkaptonuria results from the accumulation of homogentisic acid, a byproduct of tyrosine catabolism, in the urine and tissues. Oxidation of homogentisate in the urine causes it to turn dark and in the tissues results in ochronosis, which refers to the ochre color of the deposits in connective tissue, bones, and other organs. Deficiency in phenylalanine hydroxylase (choice D) results in PKU which results in severe mental retardation if not detected and treated properly. Deficiency in tyrosine aminotransferase (choice E) results in eye, skin, and neurologic symptomology. The neurologic symptoms are similar to those seen in PKU.
Question 540:
Reticulocytes control the rate of globin synthesis as a consequence of the level of heme in the cell. This prevents globin protein from being made when there are insufficient amounts of heme. Which of the following best explains the effects of heme on protein synthesis in these cells?
A. A heme-controlled phosphatase dephosphorylates cap-binding factor, which prevents recognition of globin mRNA by the ribosomes.
B. A tRNA-degrading enzyme is active in the absence of heme.
C. Heme normally activates peptidyltransfersase in reticulocytes.
D. RNA polymerase activity is decreased in reticulocytes by low heme.
E. The initiation factor eIF-2 becomes phosphorylated, reducing its level of activity.
Correct Answer: E
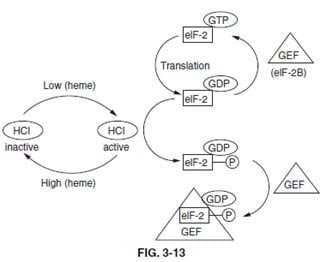
Section: Biochemistry One mechanism by which initiation of translation in eukaryotes is effected is by phosphorylation of a ser(S) residue in the alphasubunit of eIF-2. The factor eIF-2 requires activation by interaction with GTP. The energy of GTP hydrolysis is used during translational initiation, thereby allowing eIF-2 to have GDP bound instead of GTP. In order to reactivate eIF-2, the GDP must be exchanged for GTP. This requires an additional protein of the GEF family known as eIF-2B. The phosphorylated form of eIF-2, in the absence of the eIF-2B, is just as active an initiator of translation as the nonphosphorylated form. However, when eIF-2 is phosphorylated, the GDP-bound complex is stabilized and exchange for GTP is inhibited. When eIF-2 is phosphorylated, it binds eIF-2B more tightly thus slowing the rate of exchange. It is this inhibited exchange that affects the rate of initiation. Within reticulocytes the phosphorylation of eIF-2 is the result of an activity called heme-controlled inhibitor, HCI (see below figure).
The presence of HCI was first seen in an in vitro translation system derived from lysates of reticulocytes. When heme is limiting it would be a waste of energy for reticulocytes to make globin protein, since active hemoglobin could not be generated. Therefore, when the level of heme falls, HCI becomes activated, leading to the phosphorylation of eIF-2 and reduced globin synthesis. Removal of phosphate is catalyzed by a specific eIF-2 phosphatase which is unaffected by heme. There is no heme- controlled phosphatase activity (choice A) in any cell. No tRNA-specific degrading enzymes (choice B) are present in cells. There is no effect of heme levels on peptidyltransferase activity (choice C) or RNA polymerase activity (choice D).
Nowadays, the certification exams become more and more important and required by more and more enterprises when applying for a job. But how to prepare for the exam effectively? How to prepare for the exam in a short time with less efforts? How to get a ideal result and how to find the most reliable resources? Here on Vcedump.com, you will find all the answers. Vcedump.com provide not only USMLE exam questions, answers and explanations but also complete assistance on your exam preparation and certification application. If you are confused on your USMLE-STEP-1 exam preparations and USMLE certification application, do not hesitate to visit our Vcedump.com to find your solutions here.