A3-month-old infant who otherwise appeared normal during the first 2 months of life except for a bout of hyperbilirubinemia is now clearly exhibiting developmental delay. In addition, the infant's hair has become grayish and dull and there is a stubble of broken hair over the occiput and temporal regions. The facial appearance has also changed such that the infant has very pudgy cheeks, abnormal eyebrows, and sagging jowls. The occurrence of frequent convulsions was the stimulus for the parents to bring their child to the emergency room. These rapidly deteriorating symptoms are indicative of which of the following disorders?
A. Crigler-Najjar syndrome type I
B. Gilbert syndrome
C. hemochromatosis
D. Menkes disease
E. Refsum disease
Correct Answer: D
Section: Biochemistry Menkes disease is an X-linked recessive disorder that is manifest by a defect in copper absorption. This defect leads to dysfunction of numerous enzymes that need copper as a cofactor, leading to the typical symptoms observed in this patient. In fact, Menkes disease is also referred to as steely hair disease, because of the characteristic brittleness of the hair, which is easily broken. Crigler-Najjar syndrome type I (choice A) is also due to defective bilirubin metabolism as a result of a loss of UDPglucuronosyltransferase (UGT) activity. UGT is required to transfer 2 moles of glucuronic acid to bilirubin, generating bilirubindiglucuronide, which makes bilirubin much more water soluble and therefore facilitates its excretion. Crigler-Najjar syndrome results in nonhemolytic icterus (jaundice) within the first few days of life and is generally fatal during neonatal life due to severe kernicterus. Gilbert syndrome (choice B) results from a defect in bilirubin metabolism. It is typically diagnosed in young adults and is characterized by mild, chronic, and unconjugated hyperbilirubinemia without associated hemolysis. Hemochromatosis (choice C) is the term applied when organ structure and function are impaired by the presence of excess amounts of iron. The liver, heart, pancreas, skin, joints, and endocrine organs are the principal tissues affected by iron accumulation. Symptoms include cirrhosis, cardiomyopathy, arthritis, abnormal skin pigmentation, and hypogonadism, as well as diabetes mellitus. Refsum disease (choice E) results from a defect in the metabolism of phytanic acid, a plant lipid which must be oxidized by a separate pathway from that of animal fats. Cardinal symptoms include retinitis pigmentosa, peripheral neuropathy, and cerebellar ataxia.
Question 572:
Continued consumption of calories in excess of energy expenditure will eventually lead to obesity, a current major health problem in the United States. A major contributing factor in obesity is a disorder in fuel partitioning, as evidenced by a lower rate of fat oxidation in obese individuals. Which of the following situations would best explain a reduction in overall fat metabolism in these individuals?
A. An increase in the hepatic ATP/ adenosine diphosphate (ADP) ratio increases incorporation of carbon into fatty acids by causing an inhibition in acetyl-CoA oxidation in the TCA cycle.
B. Decreased hepatic gluconeogenesis which requires acetyl-CoA from fatty acid oxidation, thus fat oxidation is secondarily inhibited.
C. Increased levels of malonyl-CoA occur in these individuals leading to inhibition of carnitine palmitoyltransferase I.
D. Insulin-induced decrease in the activity of acetyl-CoA carboxylase (ACC) causing reduction in fatty acid synthesis.
E. Insulin-induced repression of fatty acid synthase (FAS) activity.
Correct Answer: C
Section: Biochemistry The oxidation of long-chain fatty acids is initiated by the sequential action of carnitine palmitoyltransferase-I (CPTI), which is located in the outer mitochondrial membrane, and carnitine palmitoyltransferase II, which is located in the inner mitochondrial, together with a carnitine- acylcarnitine translocase. Major control over the process is exerted at the level of CPTI by virtue of the unique inhibitability of this enzyme by malonyl-CoA. Thus, CPTI has a pivotal role in lipid metabolism. Obese individuals have higher overall dietary intake of lipid and the disruption in fat metabolism exacerbates the increases in malonyl-CoA, which propagates the fuel partitioning disorder. Although an increase in hepatic ATP (choice A) will lower the flux through the TCA cycle, this does not constitute a mechanism for a disruption in fuel partitioning. The rate of hepaticgluconeogenesis (choice B) does not significantly affect fat metabolism. Obese individualsactually have reduced responses to insulin (choiceD) and thus any insulin-mediated effects on ACC would be minimal. Insulin does not repress FAS activity (choice E).
Question 573:
A 9-month-old child is presented to the emergency room by his parents who report that he has been vomiting and has severe diarrhea. The episodes of vomiting began when the parents started feeding their child cow's milk. The infant exhibits signs of failing to thrive. Laboratory tests show elevated blood galactose, hypergalactosuria, metabolic acidosis, albuminuria, and hyperaminoaciduria. These clinical and laboratory findings are most consistent with which of the following disorders?
A. alkaptonuria
B. essential fructosuria
C. hereditary galactosemia
D. Menkes disease
E. von Gierke disease
Correct Answer: C
Section: Biochemistry Severe hereditary galactosemia presents in the first few months of life with symptoms that include poor feeding and associated weight loss, vomiting, diarrhea, lethargy, and hypotonia. Clinical findings will include those presented in the case. The symptoms are aggravated by consumption of cow's milk and can be resolved provided proper diagnosis is made and treatment is started early. Alkaptonuria (choice A) results from the accumulation of homogentisic acid, a by-product of tyrosine catabolism, in the urine and tissues. Oxidation of homogentisate in the urine causes it to turn dark and in the tissues results in ochronosis, which refers to the ochre color of the deposits in connective tissue, bones, and other organs. Essential fructosuria (choice B) is a benign asymptomatic metabolic disorder manifesting with alimentary hyper- fructosemia and fructosuria. Menkes disease (choice D) results from a defect in intracellular copper transport and the symptoms of the disease are caused by loss of function of copper- dependent enzymes. Symptoms include abnormal (kinky) hair and pigmentation, cerebral degeneration, failure to thrive, and skin laxity. Symptoms of von Gierke disease (choice E) result from the excessive accumulation of glycogen in liver, kidney, and intestinal mucosa. Symptoms include growth retardation, hypoglycemia, heptomegaly, hyperlipidemia, lactic acidemia, and hyperuricemia.
Question 574:
Which of the following is true with respect to the actions of the mineralocorticoids?
A. decrease carbohydrate metabolism
B. increase appearance of the secondary sex characteristics
C. increase synthesis of androgens
D. regulate aldosterone secretion
E. regulate sodium retention by the kidneys
Correct Answer: E
Section: Biochemistry The principal mineralocorticod, produced by the zona glomerulosa cells of the adrenals, is aldosterone. Synthesis of aldosterone is primarily controlled by the rennin-angiotensin system and is thus involved in control of blood pressure. Aldosterone causes sodium reabsorption by the kidneys, which in turn regulate water balance, which leads to increases in blood pressure by increasing fluid volume. Aldosterone action does not directly lead to decreased carbohydrate metabolism (choice A), is not a sex characteristic determining steroid hormone (choice B), does not result in increased synthesis of androgens (choice C), and does not regulate its own secretion (choice D).
Question 575:
When cells acquire sufficient energy such that the rate of flux through the tricarboxylic acid (TCA) cycle declines, excess acetyl-CoA that cannot be oxidized is predominantly converted into fat. In order for the carbons in mitochondrial acetyl-CoA to serve as a precursor for fat synthesis, they must be delivered to the cytosol. Which of the following represents the molecule used to transport acetyl-CoA to the cytosol?
A. acetyl-CoA
B. carnitine
C. citrate
D. beta-hydroxybutyrate
E. pyruvate
Correct Answer: C
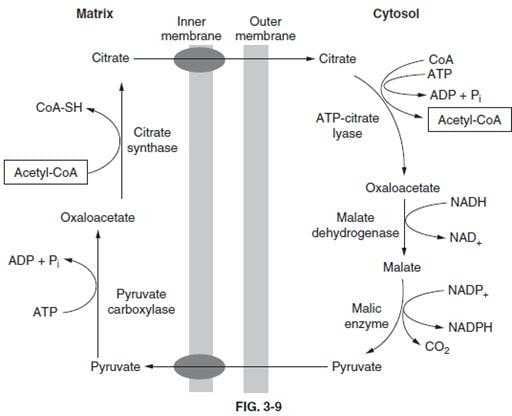
Section: Biochemistry Acetyl-CoA cannot freely diffuse across the membranes of the mitochondria, nor is there a transport mechanism to move the molecule to the cytosol. This ensures that all acetyl-CoA generated by PDH or fat oxidation will be used for energy production. However, as cellular energy demand falls, carbon atoms can be diverted into storage molecules such as glycogen and fatty acids. To move acetyl-CoA out of the mitochondria to the cytosol it must first be converted to citrate, which can be ransported by the TCA transport system (below figure).
As indicated, acetyl-CoA (choice A) is "trapped" inside the mitochondria. Carnitine (choice B) is necessary for transport of fatty acids into the mitochondria prior to their oxidation, but plays no role in acetyl-CoA transport. Ketone bodies such as beta-hydroxybutyrate (choice D) are generated in hepatic mitochodria when acetyl- CoA is in excess. However, usage of ketone bodies by nonhepatic tissues involves conversion to acetyl-CoA with the mitochondria, not the cytosol. Pyruvate (choice E) can be transported into the mitochondria but not out.
Question 576:
A 4-month-old Caucasian male infant with a temperature of 38.4°C is examined by his pediatrician. His mother indicates that he has had the fever for the past 4 days, been listless, vomiting, and has watery stools. Blood work indicates the infant is hypoglycemic but this condition does not respond to either epinephrine or glucose administration. In addition, his blood pH is slightly acidic and shows reduced bicarbonate. Other untoward blood chemistry includes elevated triglycerides, cholesterol, and liver enzymes. The child has a protruberant abdomen, thin extremities, and a doll-like face. The pediatrician suspects a specific condition and orders a liver biopsy to test for the activity of which of the following enzyme activities?
A. glucose-6-phosphatase
B. glycogen synthase
C. muscle phosphofructokinase
D. muscle phosphorylase
E. pyruvate kinase
Correct Answer: A
Section: Biochemistry Deficiency in glucose-6-phosphatase (choice A) is one cause of glycogen storage disease type I (specifically type Ia, von Gierke disease). Hallmarks of the disease are hypoglycemia, lactic acidosis, hyperuricemia, and hyperlipidemia. If symptoms do not appear until the third or fourth month they include hepatomegaly and hypoglycemic seizures. Afflicted children have a protruberant abdomen due to the massive hepatomegaly. Outward physical signs also include extremely thin extremities, short stature, and chubby doll-like faces. Liver glycogen synthase deficiency (choice B) presents with morning fatigue and ketotic hypoglycemia on fasting--both of which rapidly disappear on feeding. Symptoms can be rapidly relieved and chemical signs corrected by introducing frequent protein-rich meals and nighttime feedings of suspensions of uncooked corn starch. Adeficiency in muscle phosphofructokinase (choice C) results in glycogen storage disease type VII (Tarui disease). Clinically, the symptoms seen in Tarui disease are very similar to those seen in muscle phosphorylase deficiency (choice D), glycogen storage disease type V (McArdle disease) such as exercise-induced cramping and early fatigue. There are five clinical characteristics allowing distinction betweeen Tarui and McArdle disease: exercise intolerance is evident in childhood and more severe and is associated with nausea and vomiting; the intolerance is particularly acute following meals rich in carbohydrates; hyperuricemia is more severe; compensated hemolytic anemia is evidenced by increased serum bilirubin and reticulocyte count, and lastly; an abnormal polysaccharide is present in muscle fibers. Deficiency in PK (choice E) is the most common enzyme deficiency leading to hemolytic anemia and the disorder is characterized by lifelong episodes. The most severe deficiency will result in embryonic lethality.
Question 577:
Chromatin remodeling is associated with alterations in the transcriptional activity of genes in the region of the remodeling. Which of the following statements is most correct concerning the events of chromatin remodeling?
A. Chromatin remodeling occurs predominantly in regions enriched in CpG dinucleotides.
B. Histone acetylation tends to destabilize chromatin structure.
C. Methylation of cytosine residues induces the remodeling event.
D. Methylation of histone H1 is sufficient to stimulate remodeling.
E. Remodeling is necessary to induce the property of genomic imprinting.
Correct Answer: B
Section: Biochemistry The posttranslational modification of histone proteins has considerable effect on numerous activities at the level of the chromatin. In particular, the acetylation and/or methylation of histones in the nucleosome (H2A, H2B, H3, and H4) results in an altered stability of the 30-nm chromatin fiber as well as other higher order chromatin structure. No specific regions of remodeling have been identified relative to sequence content such as CpG islands (choice A). Methylation of cytosine residues in the DNA (choice C) has an effect on transcriptional activity but not on chromatin structure. Methylation of the linker histone, H1 (choice D) has not been demonstrated. Chromtin remodeling does not direct imprinting (choice E). Imprinting is regulated by the state of DNA methylation.
Question 578:
Analysis of a tumor cell line indicates that there is a dramatically increased level in the activity of the transcription factor E2F. Which of the following is the most likely explanation for this observation?
A. an increase in the expression of pRB resulting in increased binding of pRB to E2F
B. hypophosphorylation of pRB so that it can no longer interact with E2F
C. loss of expression of pRB which normally activates E2F
D. mutation in pRB that prevents its phosphorylation so that it cannot interact with the gene to which it normally binds and coactivates with E2F
E. mutation in the domain of pRB to which E2F binds, the consequences of which lead to constitutive E2F activity
Correct Answer: E
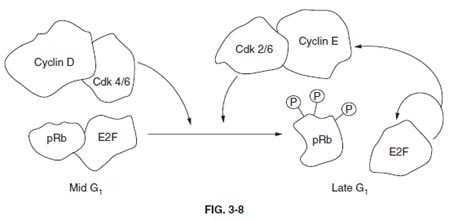
Section: Biochemistry Members of the E2F family of transcription factors play critical roles in regulating cell-cycle transit through the G1-S restriction point. The activity of E2F is regulated by interaction with the protein product of the retinoblastoma susceptibility tumor suppressor gene, pRB. Interaction of pRB and E2F occurs when pRB is in a hypophosphorylated state (below figure).
Members of the cyclin-dependent kinase family of cell-cycle regulating kinases target pRB for phosphorylation. When phosphorylated, pRB dissociates from E2F allowing E2F to enter the nucleus and transcriptionally activate genes involved in DNA synthesis, as well as activate its own transcription. Transcription of both cyclin E and CDK2 are activated by E2F. These two proteins form a complex that promotes progression through S-phase of the cell cycle and also act to keep E2F active by adding to the phosphorylation state of pRB (below figure). Thus, any defect in the ability of pRB to bind to E2F will lead to constitutive activation of DNA synthesis leading to unrestrained proliferation. None of the other options (choices AD) represent viable phenomena to account for the observed increase in E2F activity.
Question 579:
A6-month-old who is failing to thrive is brought to your clinic. Tests reveal hepatosplenomegaly, muscle weakness and atrophy, hypotonia, and decreased deep tendon reflexes. Blood tests reveal that the infant has normal glucose levels. Biopsy of the liver reveals initial stages of cirrhosis due to the accumulation of an abnormal glycogen with few branch points, whose structure resembles amylopectin. The clinical and laboratory results presented are indicative of which glycogen storage disease?
A. Andersen disease (type IV glycogen storage disease)
B. Cori or Forbes disease (type III glycogen storage disease)
C. McArdle disease (type V glycogen storage disease)
D. Tarui disease (type VII glycogen storage disease)
E. von Gierke disease (type I glycogen storage disease)
Correct Answer: A
Section: Biochemistry Andersen disease (also referred to as type IV glycogen storage disease) manifests its symptoms as a result of the accumulation of glycogen, with unbranched long outer chains in tissues. This structure of glycogen resembles that of plant amylopectin. Symptoms appear within the first year of life and lead to failure to thrive and pronounced heptosplenomegaly. Hypoglyemia is rarely seen with this disorder. Cori disease (choice B) affects both liver and muscle with accumulations of glycogen that has short outer chains (resembles limit dextrin). Symptoms include hepatomegaly, hypoglycemia, hyperlipidemia, and retarded growth. McArdle disease (choice C) usually manifests in adulthood and characteristic symptoms include exercise intolerance, muscle cramping with exercise, and myoglobinuria. The affected tissue in this disease is only skeletal muscle. Tarui disease (choice D) also affects skeletal muscle and as such manifests with clinical symptoms very similar to those of McArdle disease with the exceptions that Tarui patients also experience hemolytic anemia and myogenic hyperuricemia. Symptoms of von Gierke disease (choice E) result from the excessive accumulation of glycogen in liver, kidney, and intestinal mucosa. Symptoms include growth retardation, hypoglycemia, heptomegaly, hyperlipidemia, lactic acidemia, and hyperuricemia.
Question 580:
Gluconeogenesis is an extremely important reaction carried out in hepatocytes allowing for glucose homeostasis in the blood. The primary positive control of hepatic gluconeogenesis is exerted by which of the following?
A. high acetylcoenzyme A (acetyl-CoA) levels
B. high adenosine triphosphate (ATP) levels
C. high citrate levels
D. low ATP levels
E. low citrate levels
Correct Answer: A
Section: Biochemistry The first step in gluconeogenesis is the formation of oxaloacetate from pyruvate. The enzyme controlling this step is pyruvate carboxylase, an allosteric enzyme that does not function in the absence of its primary effector, acetyl-CoA, or closely related acyl-CoA. Thus, a high level of acetyl-CoA signals the need for more oxaloacetate. If there is a surplus of ATP, oxaloacetate will be used for gluconeogenesis. Under conditions of low ATP, oxaloacetate will be consumed in the citric acid cycle. Citrate is the primary negative effector of glycolysis and the primary positive effector of fatty acid synthesis. High levels of citrate (choice C), but not low levels (choice E), do positively affect the activity of fructose-1,6-bisphosphatase, one of the bypass enzymes of gluconeogenesis, but this is not the primary site of control, since the carbon atoms must first go through the pyruvate carboxylase reaction. Low ATP levels (choice D) would be reflected in an elevation in ADP levels, and ADP negatively affects the activity of pyruvate carboxylase. High ATP levels (choice B) are necessary in order for gluconeogenesis to proceed, and will negatively affect glycolysis at the level of PFK-1, allowing for an increased net flow of carbon into glucose. However, increased levels of ATP do not directly regulate the enzymes of gluconeogenesis.
Nowadays, the certification exams become more and more important and required by more and more enterprises when applying for a job. But how to prepare for the exam effectively? How to prepare for the exam in a short time with less efforts? How to get a ideal result and how to find the most reliable resources? Here on Vcedump.com, you will find all the answers. Vcedump.com provide not only USMLE exam questions, answers and explanations but also complete assistance on your exam preparation and certification application. If you are confused on your USMLE-STEP-1 exam preparations and USMLE certification application, do not hesitate to visit our Vcedump.com to find your solutions here.